XSEA: X-Set Enrichment Analysis (cf: ABAnnotate)#
Gene-set or gene-category enrichment analysis (GSEA or GCSEA) usually refers to analysis frameworks that test if a set of genes that you are interested in (e.g., genes found in a disease-GWAS) are found more often in some given sets of genes (e.g., marker genes of specific neural cell types) than expected under assumption of randomness. This idea was adopted to the field of neuroimaging some time ago already. The method introduced here was significantly inspired by Ben Fulcher’s paper and matlab code.
In 2023, I build a toolbox from Ben Fulcher’s code, called ABAnnotate. ABAnnotate will not be further developed, but it’s functionality is now part of NiSpace.
The Method#
“XSEA”?#
In NiSpace, we do not use the term GSEA, but generalize it to XSEA, which stands for X-Set Enrichment Analysis. We use “X” in the linear-model-sense as “predictor”. There was no reason to limit NiSpace’s set-enrichment analysis functionality to gene expression data. Instead, the XSEA methods work on any kind of input data that is organized into sets of maps. For example, we can also analyze PET maps (e.g., a serotonin set could consist of 5HT1a, 5HT2a, 5HT4, and 5HT6 maps) or neurosynth meta-analysis term maps that way (e.g., a “sensory” set could consist of maps for “visual”, “auditory”, and “somatosensory” terms).
NiSpace vs. ABAnnotate#
Flexible null model combinations
Moran randomization as the default map-based null model
Enforcing level of bilateral symmetry of the null maps to match the input map
Extending the set-enrichment method to all types of included reference data
Allen Brain Atlas mRNA data is for now provided only for genes that showed an average across-donor Spearman’s rho of at least 0.1
Many different parcellations
Works with MNI, fsaverage, and fsLR input data
Last but not least: Python as an open-source language with a large community and the likely future of neuroimaging analysis (yes, I know, I’m biased)
The following example is provided as comparison to the example in the ABAnnotate repository.
Imports#
[1]:
# load local nispace, for testing
# COMMENT THIS OUT IF YOU RUN THIS LOCALLY AFTER INSTALLING NISPACE
import sys
sys.path.append("/Users/llotter/projects/nispace/")
# general imports
import matplotlib.pyplot as plt
import pandas as pd
# import imaging functions
from nilearn.image import load_img
from nilearn.plotting import view_img
Get an input brain map#
This is a meta-analytically derived map automatically generated from the “pain” neuroscientific literature via NeuroQuery.
[2]:
# the download function is a simple function to download a file from a url and save it to a temporary directory
file_path = "neuroquery/pain.nii.gz"
# load the map into memory
effect_map = load_img(file_path)
# plot the map interactively
view_img(effect_map)
/Applications/miniforge3/envs/nsp309/lib/python3.9/site-packages/numpy/_core/fromnumeric.py:820: UserWarning: Warning: 'partition' will ignore the 'mask' of the MaskedArray.
a.partition(kth, axis=axis, kind=kind, order=order)
[2]:
Neuronal cell type marker gene sets#
For comparison with the ABAnnotate example, we will use a cell type collection: “CellTypesPsychEncodeTPM”.
fetch_collection function, or directly with the actual region-wise data via the fetch_reference function.Here, we take a look at the collection “CellTypesPsychEncodeTPM” from the “mrna” dataset.
If you want to provide your own collection, create a pandas table like the one below and pass it directly to the collection parameter.
[3]:
from nispace.datasets import fetch_collection
fetch_collection("CellTypesPsychEncodeTPM", dataset="mrna")
INFO | 15/06/25 16:09:23 | nispace: Loading integrated collection 'CellTypesPsychEncodeTPM' for dataset 'mrna'.
[3]:
| set | map | |
|---|---|---|
| 0 | Adult-Ex1 | CAMK2A |
| 1 | Adult-Ex1 | CCDC88C |
| 2 | Adult-Ex1 | CDH9 |
| 3 | Adult-Ex1 | DNM1 |
| 4 | Adult-Ex1 | GNAL |
| ... | ... | ... |
| 587 | Adult-Oligo | RNASE1 |
| 588 | Adult-Oligo | RRBP1 |
| 589 | Adult-Oligo | TF |
| 590 | Adult-Oligo | TMEM144 |
| 591 | Adult-Oligo | UGT8 |
592 rows × 2 columns
With mRNA data from the Allen Human Brain Atlas (cortex parcellation: Schaefer200), this looks like:
[4]:
from nispace.datasets import fetch_reference
# parcellation to use throughout the analysis
parcellation = "Schaefer100TianS1"
# showcase data
fetch_reference("mrna", parcellation=parcellation, collection="CellTypesPsychEncodeTPM")
INFO | 15/06/25 16:09:23 | nispace: Loading mrna maps.
INFO | 15/06/25 16:09:23 | nispace: Applying collection filter from: /Users/llotter/nispace-data/reference/mrna/collection-CellTypesPsychEncodeTPM.collect.
INFO | 15/06/25 16:09:24 | nispace: Loading and inner-merging data parcellated with 'Schaefer100' and 'TianS1'
The NiSpace "mRNA" dataset is based on Allen Human Brain Atlas (AHBA) gene expression data published in Hawrylycz et al.,
2012 (https://doi.org/10.1038/nature11405). The dataset consists of mRNA expression data from postmortem brain tissue of
six donors, mapped onto MNI or fsaverage parcels using the abagen toolbox (Markello et al., 2021, https://doi.org/10.7554/eLife.72129).
The data was extracted using abagen.get_expression_data(..., lr_mirror="bidirectional"), considering parcel hemisphere and
cortical vs. subcortical location. Only genes that showed a mean donor-to-donor Spearman correlation of > 0.1 were retained.
In addition to the two publications listed above, please cite publications associated with gene set collections as appropriate.
To ensure reproducibility, note the NiSpace commit/version: 2cb3a7f5c1cea6417fe7613465a1c503f77c87c6
- CellTypesPsychEncodeTPM Source: Lake2016 https://doi.org/10.1126/science.aaf1204
- CellTypesPsychEncodeTPM Source: Darmanis2015 https://doi.org/10.1073/pnas.1507125112
- CellTypesPsychEncodeTPM Source: Wang2018 https://doi.org/10.1126/science.aat8464
[4]:
| hemi-L_div-Vis_lab-1 | hemi-L_div-Vis_lab-2 | hemi-L_div-Vis_lab-3 | hemi-L_div-Vis_lab-4 | hemi-L_div-Vis_lab-5 | hemi-L_div-Vis_lab-6 | hemi-L_div-Vis_lab-7 | hemi-L_div-Vis_lab-8 | hemi-L_div-Vis_lab-9 | hemi-L_div-SomMot_lab-1 | ... | hemi-L_lab-PUT | hemi-L_lab-CAU | hemi-R_lab-HIP | hemi-R_lab-AMY | hemi-R_lab-pTHA | hemi-R_lab-aTHA | hemi-R_lab-NAc | hemi-R_lab-GP | hemi-R_lab-PUT | hemi-R_lab-CAU | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| set | map | |||||||||||||||||||||
| Adult-Ex1 | CAMK2A | 0.842136 | 0.826765 | 0.826967 | 0.827184 | 0.817241 | 0.822723 | 0.815382 | 0.818394 | 0.815004 | 0.819051 | ... | 0.452723 | 0.571612 | 0.792884 | 0.710187 | 0.414296 | 0.366631 | 0.485376 | 0.288345 | 0.537562 | 0.596598 |
| CDH9 | 0.742186 | 0.618615 | 0.623595 | 0.567650 | 0.546706 | 0.577347 | 0.681953 | 0.648653 | 0.656170 | 0.708508 | ... | 0.483765 | 0.509307 | 0.638481 | 0.786927 | 0.225884 | 0.266731 | 0.684596 | 0.402965 | 0.512751 | 0.539964 | |
| GNAL | 0.574698 | 0.445492 | 0.482613 | 0.427197 | 0.396329 | 0.454356 | 0.421469 | 0.467101 | 0.487929 | 0.412801 | ... | 0.761499 | 0.937351 | 0.669355 | 0.848272 | 0.777009 | 0.708689 | 0.932259 | 0.691578 | 0.719850 | 0.963261 | |
| GPR83 | 0.481428 | 0.385518 | 0.402124 | 0.335082 | 0.332499 | 0.363270 | 0.409938 | 0.360245 | 0.448235 | 0.478079 | ... | 0.407213 | 0.325885 | 0.700474 | 0.677783 | 0.243340 | 0.319397 | 0.379642 | 0.388365 | 0.434274 | 0.338194 | |
| GREB1L | 0.524661 | 0.492791 | 0.433349 | 0.499530 | 0.460485 | 0.468639 | 0.413786 | 0.436790 | 0.468180 | 0.456247 | ... | 0.697760 | 0.672921 | 0.646280 | 0.851841 | 0.478217 | 0.571359 | 0.741761 | 0.818051 | 0.610848 | 0.638883 | |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| Adult-Oligo | PLP1 | 0.496111 | 0.617112 | 0.527995 | 0.528311 | 0.550389 | 0.591656 | 0.531964 | 0.492399 | 0.555031 | 0.612572 | ... | 0.758676 | 0.655363 | 0.294118 | 0.452630 | 0.773118 | 0.683553 | 0.446722 | 0.782391 | 0.720535 | 0.704504 |
| RNASE1 | 0.482726 | 0.583546 | 0.498163 | 0.462077 | 0.527777 | 0.544846 | 0.529359 | 0.471896 | 0.515682 | 0.602738 | ... | 0.754911 | 0.719610 | 0.312573 | 0.465171 | 0.740133 | 0.661562 | 0.502950 | 0.761578 | 0.736140 | 0.771346 | |
| TF | 0.493476 | 0.601608 | 0.550798 | 0.475832 | 0.503930 | 0.596859 | 0.524158 | 0.464836 | 0.526029 | 0.621498 | ... | 0.719875 | 0.637234 | 0.269802 | 0.409345 | 0.788687 | 0.697257 | 0.445578 | 0.760539 | 0.679872 | 0.671570 | |
| TMEM144 | 0.464793 | 0.543832 | 0.462347 | 0.473897 | 0.494475 | 0.548147 | 0.490285 | 0.461037 | 0.496868 | 0.551162 | ... | 0.762305 | 0.663450 | 0.302380 | 0.481695 | 0.760091 | 0.680858 | 0.509490 | 0.794273 | 0.732716 | 0.708084 | |
| UGT8 | 0.468218 | 0.563882 | 0.481012 | 0.483897 | 0.527683 | 0.571186 | 0.510486 | 0.468666 | 0.538895 | 0.577107 | ... | 0.753372 | 0.648142 | 0.355598 | 0.497592 | 0.800845 | 0.711389 | 0.450370 | 0.788790 | 0.715404 | 0.680443 |
425 rows × 116 columns
Run the analysis#
workflow function from NiSpace. Workflow functions run a complete analysis, from data loading to plotting, with a single function call.[5]:
# set some parameters
# put into a dictionary to pass to the following functions
kwargs = {
"parcellation": parcellation, # the parcellation to use, defined above
"x_collection": "CellTypesPsychEncodeTPM", # the collection to use, here, CellTypesPsychEncodeTPM
"seed": 42, # the seed to use for the permutation analysis, for reproducibility
"n_proc": 4, # number of parallel processes to use when possible
"plot": False # dont plot, we will compare below
}
Random sampling of gene sets#
First, let’s run a “classical” set enrichment analysis with random sampling of gene sets from the whole genome dataset
[6]:
from nispace.workflows import simple_xsea
colocalization_randomgenes, p_values_randomgenes, p_fdr_values_randomgenes, nispace_object_randomgenes = simple_xsea(
y={"Pain": effect_map}, # our effect map, with the dictionary we can pass a name, can also be just the effect map
x="mrna", # Allen Human Brain Atlas mRNA data extracted with the abagen package
permute_sets=True, # permuting sets is not the default as you will see below, so we set it to True here
**kwargs
)
INFO | 15/06/25 16:09:24 | nispace: Trying to fetch background X dataset.
INFO | 15/06/25 16:09:24 | nispace: Loading mrna maps.
INFO | 15/06/25 16:09:24 | nispace: Loading and inner-merging data parcellated with 'Schaefer100' and 'TianS1'
INFO | 15/06/25 16:09:24 | nispace: Using integrated parcellation Schaefer100TianS1.
INFO | 15/06/25 16:09:24 | nispace: Loading integrated mrna dataset as X data.
INFO | 15/06/25 16:09:24 | nispace: Loading mrna maps.
INFO | 15/06/25 16:09:24 | nispace: Applying collection filter from: /Users/llotter/nispace-data/reference/mrna/collection-CellTypesPsychEncodeTPM.collect.
INFO | 15/06/25 16:09:25 | nispace: Loading and inner-merging data parcellated with 'Schaefer100' and 'TianS1'
The NiSpace "mRNA" dataset is based on Allen Human Brain Atlas (AHBA) gene expression data published in Hawrylycz et al.,
2012 (https://doi.org/10.1038/nature11405). The dataset consists of mRNA expression data from postmortem brain tissue of
six donors, mapped onto MNI or fsaverage parcels using the abagen toolbox (Markello et al., 2021, https://doi.org/10.7554/eLife.72129).
The data was extracted using abagen.get_expression_data(..., lr_mirror="bidirectional"), considering parcel hemisphere and
cortical vs. subcortical location. Only genes that showed a mean donor-to-donor Spearman correlation of > 0.1 were retained.
In addition to the two publications listed above, please cite publications associated with gene set collections as appropriate.
To ensure reproducibility, note the NiSpace commit/version: 2cb3a7f5c1cea6417fe7613465a1c503f77c87c6
- CellTypesPsychEncodeTPM Source: Lake2016 https://doi.org/10.1126/science.aaf1204
- CellTypesPsychEncodeTPM Source: Darmanis2015 https://doi.org/10.1073/pnas.1507125112
- CellTypesPsychEncodeTPM Source: Wang2018 https://doi.org/10.1126/science.aat8464
INFO | 15/06/25 16:09:25 | nispace: *** NiSpace.fit() - Data extraction and preparation. ***
INFO | 15/06/25 16:09:25 | nispace: Loading cortex parcellation 'Schaefer100' in 'MNI152NLin2009cAsym' space.
INFO | 15/06/25 16:09:25 | nispace: Loading subcortex parcellation 'TianS1' in 'MNI152NLin2009cAsym' space.
INFO | 15/06/25 16:09:25 | nispace: Merging to cortex-subcortex parcellation 'Schaefer100TianS1'.
INFO | 15/06/25 16:09:25 | nispace: Distance matrices for merged parcellations are currently not available. Returning None.
INFO | 15/06/25 16:09:25 | nispace: Loaded integrated parcellation with pre-calculated distance matrix.
INFO | 15/06/25 16:09:25 | nispace: Checking input data for 'x' (should be, e.g., PET data):
INFO | 15/06/25 16:09:25 | nispace: Input type: DataFrame, assuming parcellated data with shape (n_files/subjects/etc, n_parcels).
INFO | 15/06/25 16:09:25 | nispace: Got 'x' data for 425 x 116 parcels.
INFO | 15/06/25 16:09:25 | nispace: Checking input data for 'y' (should be, e.g., subject data):
INFO | 15/06/25 16:09:25 | nispace: Input type: dict, assuming (img_name, img) pairs for imaging data.
INFO | 15/06/25 16:09:25 | nispace: Parcellating imaging data.
INFO | 15/06/25 16:09:29 | nispace: Combined across images, 0 parcel(s) had only background intensity and were set to nan ([]).
INFO | 15/06/25 16:09:29 | nispace: Got 'y' data for 1 x 116 parcels.
INFO | 15/06/25 16:09:29 | nispace: Z-standardizing 'X' data.
INFO | 15/06/25 16:09:29 | nispace: *** NiSpace.colocalize() - Estimating X & Y colocalizations. ***
INFO | 15/06/25 16:09:29 | nispace: Running 'spearman' colocalization.
INFO | 15/06/25 16:09:29 | nispace: Will perform X-set enrichment analysis (XSEA).
INFO | 15/06/25 16:09:29 | nispace: Using 24 sets with between 2 and 59 samples. Aggregating within-set colocalizations with: mean.
INFO | 15/06/25 16:09:32 | nispace: *** NiSpace.permute() - Estimate exact non-parametric p values. ***
INFO | 15/06/25 16:09:32 | nispace: Permutation of: X sets.
INFO | 15/06/25 16:09:32 | nispace: Loading observed colocalizations (method = 'spearman').
INFO | 15/06/25 16:09:32 | nispace: Generating permuted X sets.
INFO | 15/06/25 16:09:32 | nispace: Will use 9562 provided background maps.
INFO | 15/06/25 16:09:32 | nispace: Z-standardizing X background maps.
INFO | 15/06/25 16:09:46 | nispace: Calculating exact p-values (tails = {'rho': 'two'}).
INFO | 15/06/25 16:09:46 | nispace: *** NiSpace.correct_p() - Correct p values for multiple comparisons. ***
INFO | 15/06/25 16:09:46 | nispace: Returning colocalizations:
| METHOD | XSEA | X_REDUCTION | Y_TRANSFORM |
| spearman | True | False | False |
INFO | 15/06/25 16:09:46 | nispace: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | NORM | X_REDUCTION | Y_TRANSFORM |
| spearman | sets | True | None | False | False | False |
INFO | 15/06/25 16:09:46 | nispace: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | NORM | X_REDUCTION | Y_TRANSFORM |
| spearman | sets | True | fdrbh | False | False | False |
Permutation of the input map#
Now, let’s run a set-enrichment analysis, but do not randomize the gene sets and rather “permute” the input map (not actually permutation but sampling of null maps with similar spatial structure). This is the default setting in NiSpace adopted from ABAnnotate and Fulcher’s paper.
[7]:
colocalization_randominput, p_values_randominput, p_fdr_values_randominput, nispace_object_randominput = simple_xsea(
y={"Pain": effect_map}, # our effect map, with the dictionary we can pass a name, can also be just the effect map
x="mrna", # Allen Human Brain Atlas mRNA data extracted with the abagen package
**kwargs
)
INFO | 15/06/25 16:09:46 | nispace: Using integrated parcellation Schaefer100TianS1.
INFO | 15/06/25 16:09:46 | nispace: Loading integrated mrna dataset as X data.
INFO | 15/06/25 16:09:46 | nispace: Loading mrna maps.
INFO | 15/06/25 16:09:46 | nispace: Applying collection filter from: /Users/llotter/nispace-data/reference/mrna/collection-CellTypesPsychEncodeTPM.collect.
INFO | 15/06/25 16:09:47 | nispace: Loading and inner-merging data parcellated with 'Schaefer100' and 'TianS1'
The NiSpace "mRNA" dataset is based on Allen Human Brain Atlas (AHBA) gene expression data published in Hawrylycz et al.,
2012 (https://doi.org/10.1038/nature11405). The dataset consists of mRNA expression data from postmortem brain tissue of
six donors, mapped onto MNI or fsaverage parcels using the abagen toolbox (Markello et al., 2021, https://doi.org/10.7554/eLife.72129).
The data was extracted using abagen.get_expression_data(..., lr_mirror="bidirectional"), considering parcel hemisphere and
cortical vs. subcortical location. Only genes that showed a mean donor-to-donor Spearman correlation of > 0.1 were retained.
In addition to the two publications listed above, please cite publications associated with gene set collections as appropriate.
To ensure reproducibility, note the NiSpace commit/version: 2cb3a7f5c1cea6417fe7613465a1c503f77c87c6
- CellTypesPsychEncodeTPM Source: Lake2016 https://doi.org/10.1126/science.aaf1204
- CellTypesPsychEncodeTPM Source: Darmanis2015 https://doi.org/10.1073/pnas.1507125112
- CellTypesPsychEncodeTPM Source: Wang2018 https://doi.org/10.1126/science.aat8464
INFO | 15/06/25 16:09:47 | nispace: *** NiSpace.fit() - Data extraction and preparation. ***
INFO | 15/06/25 16:09:47 | nispace: Loading cortex parcellation 'Schaefer100' in 'MNI152NLin2009cAsym' space.
INFO | 15/06/25 16:09:47 | nispace: Loading subcortex parcellation 'TianS1' in 'MNI152NLin2009cAsym' space.
INFO | 15/06/25 16:09:47 | nispace: Merging to cortex-subcortex parcellation 'Schaefer100TianS1'.
INFO | 15/06/25 16:09:47 | nispace: Distance matrices for merged parcellations are currently not available. Returning None.
INFO | 15/06/25 16:09:47 | nispace: Loaded integrated parcellation with pre-calculated distance matrix.
INFO | 15/06/25 16:09:47 | nispace: Checking input data for 'x' (should be, e.g., PET data):
INFO | 15/06/25 16:09:47 | nispace: Input type: DataFrame, assuming parcellated data with shape (n_files/subjects/etc, n_parcels).
INFO | 15/06/25 16:09:47 | nispace: Got 'x' data for 425 x 116 parcels.
INFO | 15/06/25 16:09:47 | nispace: Checking input data for 'y' (should be, e.g., subject data):
INFO | 15/06/25 16:09:47 | nispace: Input type: dict, assuming (img_name, img) pairs for imaging data.
INFO | 15/06/25 16:09:47 | nispace: Parcellating imaging data.
INFO | 15/06/25 16:09:48 | nispace: Combined across images, 0 parcel(s) had only background intensity and were set to nan ([]).
INFO | 15/06/25 16:09:48 | nispace: Got 'y' data for 1 x 116 parcels.
INFO | 15/06/25 16:09:48 | nispace: Z-standardizing 'X' data.
INFO | 15/06/25 16:09:48 | nispace: *** NiSpace.colocalize() - Estimating X & Y colocalizations. ***
INFO | 15/06/25 16:09:48 | nispace: Running 'spearman' colocalization.
INFO | 15/06/25 16:09:48 | nispace: Will perform X-set enrichment analysis (XSEA).
INFO | 15/06/25 16:09:48 | nispace: Using 24 sets with between 2 and 59 samples. Aggregating within-set colocalizations with: mean.
INFO | 15/06/25 16:09:48 | nispace: *** NiSpace.permute() - Estimate exact non-parametric p values. ***
INFO | 15/06/25 16:09:48 | nispace: Permutation of: Y maps.
INFO | 15/06/25 16:09:48 | nispace: Loading observed colocalizations (method = 'spearman').
INFO | 15/06/25 16:09:48 | nispace: Generating permuted Y maps.
INFO | 15/06/25 16:09:48 | nispace: No null maps found.
INFO | 15/06/25 16:09:48 | nispace: Generating null maps (n = 10000, null_method = 'moran').
INFO | 15/06/25 16:09:48 | nispace: Null map generation: Assuming n = 1 data vector(s) for n = 116 parcels.
INFO | 15/06/25 16:09:48 | nispace: Loaded parcellation (parc_space = 'MNI152NLin2009cAsym').
INFO | 15/06/25 16:09:48 | nispace: Downsampling volumetric parcellation to voxelsize of 2 for distance matrix generation.
INFO | 15/06/25 16:09:49 | nispace: Estimating euclidean distance matrix between 116 volumetric parcels.
/Applications/miniforge3/envs/nsp309/lib/python3.9/site-packages/joblib/externals/loky/process_executor.py:752: UserWarning: A worker stopped while some jobs were given to the executor. This can be caused by a too short worker timeout or by a memory leak.
warnings.warn(
INFO | 15/06/25 16:09:57 | nispace: Generating null data separately for left and right hemisphere.
INFO | 15/06/25 16:10:00 | nispace: Left-to-right mirroring null maps (using left-to-right mapping).
INFO | 15/06/25 16:10:00 | nispace: Matching interhemispheric correlation. Observed correlation: 0.86
INFO | 15/06/25 16:10:03 | nispace: Null data generation finished.
INFO | 15/06/25 16:10:03 | nispace: Running X Set Enrichment Analysis (XSEA) without set permutation.
INFO | 15/06/25 16:10:19 | nispace: Calculating exact p-values (tails = {'rho': 'two'}).
INFO | 15/06/25 16:10:19 | nispace: *** NiSpace.correct_p() - Correct p values for multiple comparisons. ***
INFO | 15/06/25 16:10:19 | nispace: Returning colocalizations:
| METHOD | XSEA | X_REDUCTION | Y_TRANSFORM |
| spearman | True | False | False |
INFO | 15/06/25 16:10:19 | nispace: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | NORM | X_REDUCTION | Y_TRANSFORM |
| spearman | ymaps | True | None | False | False | False |
INFO | 15/06/25 16:10:19 | nispace: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | NORM | X_REDUCTION | Y_TRANSFORM |
| spearman | ymaps | True | fdrbh | False | False | False |
Inspect the results#
The workflow function returns dataframes with colocalization values, p values, and FDR-corrected p values. Let’s take a look at the p values.
[8]:
# Concatenate p values from both methods
p_values = pd.DataFrame(
{
"randomgenes": p_values_randomgenes.loc["Pain"],
"randominput": p_values_randominput.loc["Pain"],
}
)
# Display, sorted by the input map permutation results
p_values.sort_values(by="randominput")
[8]:
| randomgenes | randominput | |
|---|---|---|
| Adult-Ex8 | 0.0014 | 0.0056 |
| Dev-replicating | 0.0456 | 0.0152 |
| Adult-OPC | 0.0001 | 0.0276 |
| Adult-Micro | 0.0001 | 0.0580 |
| Adult-Ex7 | 0.1162 | 0.0604 |
| Adult-Ex6 | 0.0006 | 0.0906 |
| Adult-Oligo | 0.0032 | 0.0934 |
| Adult-Endo | 0.0001 | 0.1850 |
| Adult-Astro | 0.0004 | 0.2574 |
| Adult-Ex3 | 0.0086 | 0.2756 |
| Dev-quiescent | 0.3082 | 0.3680 |
| Adult-In2 | 0.6994 | 0.3924 |
| Adult-In1 | 0.3842 | 0.4754 |
| Adult-In5 | 0.4862 | 0.6524 |
| Adult-Ex2 | 0.7084 | 0.6854 |
| Adult-OtherNeuron | 0.7222 | 0.7662 |
| Adult-Ex4 | 0.4942 | 0.8630 |
| Adult-In3 | 0.8662 | 0.8866 |
| Adult-In6 | 0.8304 | 0.9022 |
| Adult-Ex1 | 0.9192 | 0.9080 |
| Adult-Ex5 | 0.6488 | 0.9222 |
| Adult-In4 | 0.9684 | 0.9864 |
| Adult-In7 | 0.9676 | 0.9948 |
| Adult-In8 | 0.9994 | 0.9994 |
However, we can also see two more things:
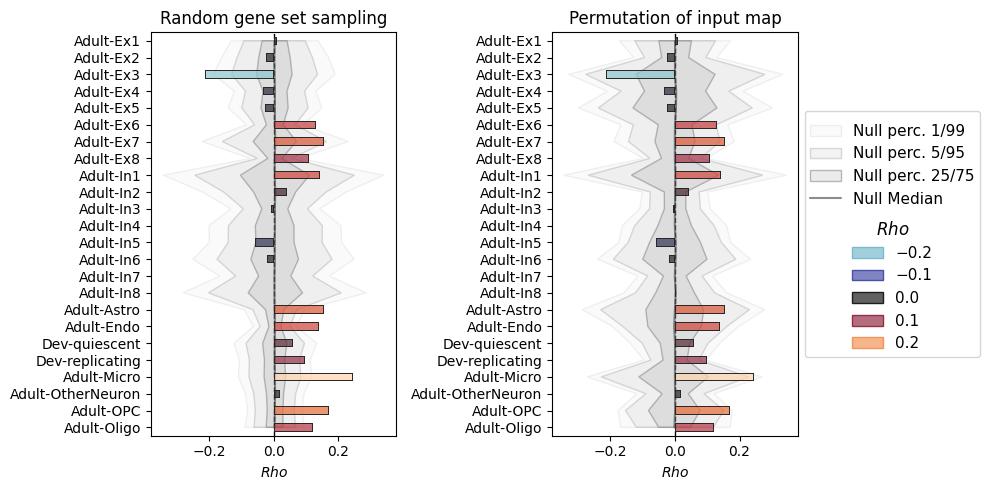
For the random gene set sampling, much more maps turn out to be significant than for the input null map method. This might indeed be an indication of inflated false positives! The initially cited paper by Fulcher et al., 2021 suggested this method to be affected by within-set coexpression patterns. The input null map method cannot have this problem, as the gene sets remain unchanged during the null runs.
The results seem somewhat more conservative than in the ABAnnotate example. As listed above, there are many implementation differences, and I would generally suggest to trust the much more advanced NiSpace implementation.
Plot side by side#
We have an integrated plotting method for the NiSpace objects that were returned by the workflow functions.
[9]:
# create figure to host both plots
fig, axes = plt.subplots(1, 2, figsize=(10, 5), sharey=True, sharex=True)
# plot both plots
nispace_object_randomgenes.plot(ax=axes[0], show=False, title="Random gene set sampling", fig=fig)
nispace_object_randominput.plot(ax=axes[1], show=False, title="Permutation of input map", fig=fig)
# hide legend of first plot
axes[0].legend_.set_visible(False)
# adjust layout
fig.tight_layout()
INFO | 15/06/25 16:10:19 | nispace: *** NiSpace.plot() - Plot colocalization results. ***
INFO | 15/06/25 16:10:20 | nispace: Creating categorical plot for method spearman, colocalization stat rho.
INFO | 15/06/25 16:10:20 | nispace: *** NiSpace.plot() - Plot colocalization results. ***
INFO | 15/06/25 16:10:21 | nispace: Creating categorical plot for method spearman, colocalization stat rho.
Extra: Colocalization methods#
We have partial least squares ("pls") and principal component regression ("pcr") as options.
Let’s take a look at the results using PLS (default setting: n = 1 component).
[10]:
colocalization_randomgenes_pls, p_values_randomgenes_pls, p_fdr_values_randomgenes_pls, _ = simple_xsea(
y={"Pain": effect_map},
x="mrna",
colocalization_method="pls",
permute_sets=True,
**kwargs
)
INFO | 15/06/25 16:10:21 | nispace: Trying to fetch background X dataset.
INFO | 15/06/25 16:10:21 | nispace: Loading mrna maps.
INFO | 15/06/25 16:10:21 | nispace: Loading and inner-merging data parcellated with 'Schaefer100' and 'TianS1'
INFO | 15/06/25 16:10:21 | nispace: Using integrated parcellation Schaefer100TianS1.
INFO | 15/06/25 16:10:21 | nispace: Loading integrated mrna dataset as X data.
INFO | 15/06/25 16:10:21 | nispace: Loading mrna maps.
INFO | 15/06/25 16:10:21 | nispace: Applying collection filter from: /Users/llotter/nispace-data/reference/mrna/collection-CellTypesPsychEncodeTPM.collect.
INFO | 15/06/25 16:10:22 | nispace: Loading and inner-merging data parcellated with 'Schaefer100' and 'TianS1'
The NiSpace "mRNA" dataset is based on Allen Human Brain Atlas (AHBA) gene expression data published in Hawrylycz et al.,
2012 (https://doi.org/10.1038/nature11405). The dataset consists of mRNA expression data from postmortem brain tissue of
six donors, mapped onto MNI or fsaverage parcels using the abagen toolbox (Markello et al., 2021, https://doi.org/10.7554/eLife.72129).
The data was extracted using abagen.get_expression_data(..., lr_mirror="bidirectional"), considering parcel hemisphere and
cortical vs. subcortical location. Only genes that showed a mean donor-to-donor Spearman correlation of > 0.1 were retained.
In addition to the two publications listed above, please cite publications associated with gene set collections as appropriate.
To ensure reproducibility, note the NiSpace commit/version: 2cb3a7f5c1cea6417fe7613465a1c503f77c87c6
- CellTypesPsychEncodeTPM Source: Lake2016 https://doi.org/10.1126/science.aaf1204
- CellTypesPsychEncodeTPM Source: Darmanis2015 https://doi.org/10.1073/pnas.1507125112
- CellTypesPsychEncodeTPM Source: Wang2018 https://doi.org/10.1126/science.aat8464
INFO | 15/06/25 16:10:22 | nispace: *** NiSpace.fit() - Data extraction and preparation. ***
INFO | 15/06/25 16:10:22 | nispace: Loading cortex parcellation 'Schaefer100' in 'MNI152NLin2009cAsym' space.
INFO | 15/06/25 16:10:22 | nispace: Loading subcortex parcellation 'TianS1' in 'MNI152NLin2009cAsym' space.
INFO | 15/06/25 16:10:22 | nispace: Merging to cortex-subcortex parcellation 'Schaefer100TianS1'.
INFO | 15/06/25 16:10:22 | nispace: Distance matrices for merged parcellations are currently not available. Returning None.
INFO | 15/06/25 16:10:22 | nispace: Loaded integrated parcellation with pre-calculated distance matrix.
INFO | 15/06/25 16:10:22 | nispace: Checking input data for 'x' (should be, e.g., PET data):
INFO | 15/06/25 16:10:22 | nispace: Input type: DataFrame, assuming parcellated data with shape (n_files/subjects/etc, n_parcels).
INFO | 15/06/25 16:10:22 | nispace: Got 'x' data for 425 x 116 parcels.
INFO | 15/06/25 16:10:22 | nispace: Checking input data for 'y' (should be, e.g., subject data):
INFO | 15/06/25 16:10:22 | nispace: Input type: dict, assuming (img_name, img) pairs for imaging data.
INFO | 15/06/25 16:10:22 | nispace: Parcellating imaging data.
INFO | 15/06/25 16:10:23 | nispace: Combined across images, 0 parcel(s) had only background intensity and were set to nan ([]).
INFO | 15/06/25 16:10:23 | nispace: Got 'y' data for 1 x 116 parcels.
INFO | 15/06/25 16:10:23 | nispace: Z-standardizing 'X' data.
INFO | 15/06/25 16:10:23 | nispace: *** NiSpace.colocalize() - Estimating X & Y colocalizations. ***
INFO | 15/06/25 16:10:23 | nispace: Running 'pls' colocalization.
INFO | 15/06/25 16:10:23 | nispace: Will perform X-set enrichment analysis (XSEA).
INFO | 15/06/25 16:10:23 | nispace: Using 24 sets with between 2 and 59 samples. Aggregating within-set colocalizations with: mean.
INFO | 15/06/25 16:10:23 | nispace: *** NiSpace.permute() - Estimate exact non-parametric p values. ***
INFO | 15/06/25 16:10:23 | nispace: Permutation of: X sets.
INFO | 15/06/25 16:10:23 | nispace: Loading observed colocalizations (method = 'pls').
INFO | 15/06/25 16:10:23 | nispace: Generating permuted X sets.
INFO | 15/06/25 16:10:23 | nispace: Will use 9562 provided background maps.
INFO | 15/06/25 16:10:23 | nispace: Z-standardizing X background maps.
INFO | 15/06/25 16:10:53 | nispace: Calculating exact p-values (tails = {'r2': 'upper', 'beta': 'two'}).
INFO | 15/06/25 16:10:54 | nispace: *** NiSpace.correct_p() - Correct p values for multiple comparisons. ***
INFO | 15/06/25 16:10:54 | nispace: Returning colocalizations:
| METHOD | XSEA | X_REDUCTION | Y_TRANSFORM |
| pls | True | False | False |
INFO | 15/06/25 16:10:54 | nispace: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | NORM | X_REDUCTION | Y_TRANSFORM |
| pls | sets | True | None | False | False | False |
INFO | 15/06/25 16:10:54 | nispace: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | NORM | X_REDUCTION | Y_TRANSFORM |
| pls | sets | True | fdrbh | False | False | False |
[11]:
p_values_randomgenes_pls["beta"].loc["Pain"].sort_values()
[11]:
Adult-Endo 0.0001
Adult-Ex8 0.0034
Adult-Micro 0.0110
Adult-OPC 0.0156
Adult-Oligo 0.0202
Adult-Ex6 0.1014
Adult-Astro 0.1274
Adult-Ex4 0.1374
Adult-Ex7 0.1740
Adult-In3 0.1796
Adult-Ex1 0.2602
Adult-Ex3 0.2690
Adult-Ex5 0.2790
Dev-replicating 0.4708
Adult-In6 0.6770
Adult-In4 0.6938
Adult-OtherNeuron 0.7052
Adult-In7 0.7660
Adult-In1 0.8462
Adult-Ex2 0.8580
Adult-In2 0.9572
Dev-quiescent 0.9782
Adult-In5 0.9838
Adult-In8 0.9978
Name: Pain, dtype: float32
Extra: Other mRNA expression dataset#
The data is reconstructed on the cortex, so we will use the Schaefer100 parcellation.
[12]:
colocalization_randomgenes_plsmagicc, p_values_randomgenes_plsmagicc, p_fdr_values_randomgenes_plsmagicc, _ = simple_xsea(
y={"Pain": effect_map},
x="magicc",
colocalization_method="pls",
permute_sets=True,
**kwargs | {"parcellation": "Schaefer100"}
)
INFO | 15/06/25 16:10:54 | nispace: Trying to fetch background X dataset.
INFO | 15/06/25 16:10:54 | nispace: Loading magicc maps.
INFO | 15/06/25 16:10:54 | nispace: Loading data parcellated with 'Schaefer100'
INFO | 15/06/25 16:10:54 | nispace: Using integrated parcellation Schaefer100.
INFO | 15/06/25 16:10:54 | nispace: Loading integrated magicc dataset as X data.
INFO | 15/06/25 16:10:54 | nispace: Loading magicc maps.
INFO | 15/06/25 16:10:54 | nispace: Applying collection filter from: /Users/llotter/nispace-data/reference/magicc/collection-CellTypesPsychEncodeTPM.collect.
INFO | 15/06/25 16:10:54 | nispace: Loading data parcellated with 'Schaefer100'
The NiSpace "magicc" dataset is based on Allen Human Brain Atlas (AHBA) gene expression data published in Hawrylycz et al.,
2012 (https://doi.org/10.1038/nature11405). The dataset consists of mRNA expression data from postmortem brain tissue of
six donors, mapped onto continuous fsLR space as described in Wagstyl et al., 2024 (https://doi.org/10.7554/eLife.86933.1),
and parcellated with integrated parcellation. Only genes that showed a high reproducibility (>= 0.5, see paper) were retained.
In addition to the two publications listed above, please cite publications associated with gene set collections as appropriate.
To ensure reproducibility, note the NiSpace commit/version: 2cb3a7f5c1cea6417fe7613465a1c503f77c87c6
- CellTypesPsychEncodeTPM Source: Lake2016 https://doi.org/10.1126/science.aaf1204
- CellTypesPsychEncodeTPM Source: Darmanis2015 https://doi.org/10.1073/pnas.1507125112
- CellTypesPsychEncodeTPM Source: Wang2018 https://doi.org/10.1126/science.aat8464
INFO | 15/06/25 16:10:54 | nispace: *** NiSpace.fit() - Data extraction and preparation. ***
INFO | 15/06/25 16:10:54 | nispace: Loading cortex parcellation 'Schaefer100' in 'MNI152NLin2009cAsym' space.
INFO | 15/06/25 16:10:54 | nispace: Loaded integrated parcellation with pre-calculated distance matrix.
INFO | 15/06/25 16:10:54 | nispace: Checking input data for 'x' (should be, e.g., PET data):
INFO | 15/06/25 16:10:54 | nispace: Input type: DataFrame, assuming parcellated data with shape (n_files/subjects/etc, n_parcels).
INFO | 15/06/25 16:10:54 | nispace: Got 'x' data for 445 x 100 parcels.
INFO | 15/06/25 16:10:54 | nispace: Checking input data for 'y' (should be, e.g., subject data):
INFO | 15/06/25 16:10:54 | nispace: Input type: dict, assuming (img_name, img) pairs for imaging data.
INFO | 15/06/25 16:10:54 | nispace: Parcellating imaging data.
INFO | 15/06/25 16:10:55 | nispace: Combined across images, 0 parcel(s) had only background intensity and were set to nan ([]).
INFO | 15/06/25 16:10:55 | nispace: Got 'y' data for 1 x 100 parcels.
INFO | 15/06/25 16:10:55 | nispace: Z-standardizing 'X' data.
INFO | 15/06/25 16:10:55 | nispace: *** NiSpace.colocalize() - Estimating X & Y colocalizations. ***
INFO | 15/06/25 16:10:55 | nispace: Running 'pls' colocalization.
INFO | 15/06/25 16:10:55 | nispace: Will perform X-set enrichment analysis (XSEA).
INFO | 15/06/25 16:10:55 | nispace: Using 24 sets with between 2 and 51 samples. Aggregating within-set colocalizations with: mean.
INFO | 15/06/25 16:10:55 | nispace: *** NiSpace.permute() - Estimate exact non-parametric p values. ***
INFO | 15/06/25 16:10:55 | nispace: Permutation of: X sets.
INFO | 15/06/25 16:10:55 | nispace: Loading observed colocalizations (method = 'pls').
INFO | 15/06/25 16:10:55 | nispace: Generating permuted X sets.
INFO | 15/06/25 16:10:55 | nispace: Will use 11146 provided background maps.
INFO | 15/06/25 16:10:55 | nispace: Z-standardizing X background maps.
INFO | 15/06/25 16:11:25 | nispace: Calculating exact p-values (tails = {'r2': 'upper', 'beta': 'two'}).
INFO | 15/06/25 16:11:25 | nispace: *** NiSpace.correct_p() - Correct p values for multiple comparisons. ***
INFO | 15/06/25 16:11:25 | nispace: Returning colocalizations:
| METHOD | XSEA | X_REDUCTION | Y_TRANSFORM |
| pls | True | False | False |
INFO | 15/06/25 16:11:25 | nispace: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | NORM | X_REDUCTION | Y_TRANSFORM |
| pls | sets | True | None | False | False | False |
INFO | 15/06/25 16:11:25 | nispace: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | NORM | X_REDUCTION | Y_TRANSFORM |
| pls | sets | True | fdrbh | False | False | False |
Similar results reassure us of the robustness of our findings.
[13]:
p_values_randomgenes_plsmagicc["beta"].loc["Pain"].sort_values()
[13]:
Adult-Ex8 0.0002
Adult-Endo 0.0042
Dev-replicating 0.0336
Adult-Ex4 0.0362
Adult-Ex3 0.0372
Adult-Ex6 0.0384
Adult-Oligo 0.0396
Adult-Ex5 0.0990
Adult-Astro 0.1078
Adult-In4 0.1254
Adult-OPC 0.1742
Adult-OtherNeuron 0.2674
Adult-In7 0.2678
Adult-Micro 0.2874
Adult-In5 0.4460
Adult-Ex1 0.4966
Adult-In6 0.5356
Adult-In3 0.5696
Adult-In1 0.6626
Adult-Ex7 0.7564
Adult-In8 0.8386
Adult-In2 0.8938
Dev-quiescent 0.8966
Adult-Ex2 0.9008
Name: Pain, dtype: float32