Getting started: the NiSpace object
This notebook introduces the core NiSpace API through a complete, step-by-step colocalization analysis. If you haven’t already, have a look at the Spatial Colocalization notebook for the conceptual background.
We’ll use a NeuroQuery meta-analytic map for “pain” as our input and test whether it colocalizes with a set of PET-derived neurotransmitter receptor maps. This is a clean, self-contained example with a nice biological story.
[2]:
# make progress bars render as text in the docs
import tqdm.notebook
tqdm.notebook.tqdm = tqdm.tqdm
The input map: a NeuroQuery pain map
We need a brain map to start with. I downloaded a meta-analytic map from NeuroQuery — a tool that generates brain activation maps from text queries against the neuroimaging literature. The map for “pain” is a z-score image reflecting how consistently the pain literature reports activation across brain regions.
Technically this is not a T-map from a single study, but it serves our purpose perfectly: it’s a plausible spatial pattern of pain-related brain activity, and it lives in MNI space like all our reference data.
[3]:
from nispace.io import load_img
from nispace.plotting import brainplot
# load the pain map
pain_map = load_img("neuroquery/pain.nii.gz")
# plot it
brainplot(pain_map, title="Pain (NeuroQuery)")
WARNING | 20/07/26 18:19:56 | nispace.plotting: Brain plotting in NiSpace is experimental. If things look off, feel free to raise a GitHub issue!
[3]:
(<Figure size 720x180 with 6 Axes>, [<Axes: >])
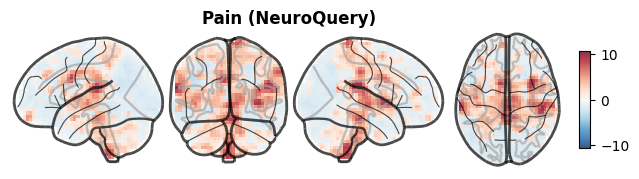
The map shows the expected pattern: activation in regions associated with pain processing, including the insula, anterior cingulate cortex, and somatosensory cortex.
Reference data: PET receptor maps
For our reference maps, we’ll use NiSpace’s curated PET receptor dataset. This collection contains group-average receptor density maps for a wide range of neurotransmitter receptors and transporters, derived from PET studies in healthy subjects.
We fetch the data already parcellated into our target parcellation. This is much faster than loading the volumetric images and parcellating them on the fly — NiSpace ships pre-parcellated versions of all reference datasets.
Throughout this series we use Yan200 as the default parcellation. Yan et al. (2023) introduced this as an updated, homotopic version of the widely used Schaefer parcellation: each left-hemisphere parcel has an exact right-hemisphere counterpart of the same shape, which plays nicely with the spin-based null model methods used in permutation testing. We use the 17-network Kong variant. See the Parcellations page for more options.
[4]:
from nispace.datasets import fetch_reference
# fetch the PET maps, pre-parcellated into Yan200
# collection="UniqueTracers" picks one representative tracer per receptor target
pet_maps = fetch_reference(
"pet",
parcellation="Yan200",
collection="UniqueTracers",
print_references=False # suppress the long reference list for now
)
print(f"PET DataFrame: {pet_maps.shape[0]} maps x {pet_maps.shape[1]} parcels")
pet_maps.head(3)
INFO | 20/07/26 18:19:56 | nispace.datasets: Loading pet maps.
INFO | 20/07/26 18:19:56 | nispace.datasets: Loading integrated collection 'UniqueTracers' for dataset 'pet'.
INFO | 20/07/26 18:19:56 | nispace.datasets: Filtering maps by collection.
INFO | 20/07/26 18:19:56 | nispace.datasets: Loading data parcellated with 'Yan200'
PET DataFrame: 29 maps x 200 parcels
[4]:
| hemi-L_div-DefaultC_lab-IPL | hemi-L_div-DefaultC_lab-PHC | hemi-L_div-DefaultB_lab-IPL+1 | hemi-L_div-DefaultB_lab-IPL+2 | hemi-L_div-DefaultB_lab-PFCd+1 | hemi-L_div-DefaultB_lab-PFCd+2 | hemi-L_div-DefaultB_lab-PFCl | hemi-L_div-DefaultB_lab-PFCm | hemi-L_div-DefaultB_lab-PFCv+1 | hemi-L_div-DefaultB_lab-PFCv+2 | ... | hemi-R_div-VisualB_lab-ExStrSup | hemi-R_div-VisualB_lab-Striate+1 | hemi-R_div-VisualB_lab-Striate+2 | hemi-R_div-VisualA_lab-ExStr+1 | hemi-R_div-VisualA_lab-ExStr+2 | hemi-R_div-VisualA_lab-ExStr+3 | hemi-R_div-VisualA_lab-ExStr+4 | hemi-R_div-VisualA_lab-SPL | hemi-R_div-VisualA_lab-TempOcc+1 | hemi-R_div-VisualA_lab-TempOcc+2 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| set | map | |||||||||||||||||||||
| General | target-CMRglu_tracer-fdg_n-20_dx-hc_pub-castrillon2023 | 0.636817 | 0.540483 | 0.682817 | 0.728153 | 0.578478 | 0.529284 | 0.693211 | 0.665677 | 0.723623 | 0.703104 | ... | 0.783619 | 0.794409 | 0.695514 | 0.627114 | 0.650671 | 0.588715 | 0.643129 | 0.612235 | 0.664404 | 0.627583 |
| target-rCPS_tracer-leucine_n-42_dx-hc_pub-smith2023 | 0.634173 | 0.438008 | 0.611914 | 0.624071 | 0.428779 | 0.431085 | 0.541520 | 0.513382 | 0.596148 | 0.499352 | ... | 0.655291 | 0.765224 | 0.706545 | 0.641496 | 0.645077 | 0.593723 | 0.597942 | 0.512976 | 0.633523 | 0.604931 | |
| target-SV2A_tracer-ucbj_n-76_dx-hc_pub-finnema2016 | 0.672142 | 0.540954 | 0.680068 | 0.686451 | 0.503211 | 0.541792 | 0.565813 | 0.666088 | 0.641296 | 0.617081 | ... | 0.658140 | 0.661806 | 0.584824 | 0.680765 | 0.616637 | 0.601831 | 0.670417 | 0.593934 | 0.696135 | 0.666245 |
3 rows × 200 columns
The DataFrame has a two-level index: the first level is the neurotransmitter system (“set”), the second is the full tracer identifier. These sets are used later for X-Set Enrichment Analysis (XSEA notebook) — for now we just treat the whole thing as a collection of maps.
Initializing the NiSpace object
NiSpace follows an object-oriented design. We first create a NiSpace instance, passing all the data and configuration options. Processing happens later when we call the analysis methods.
The two central arguments are:
x: the reference maps (here: PET receptor maps)y: the input map(s) we want to analyze (here: the pain map)
We also specify the parcellation, which tells NiSpace how to parcellate y (since it’s a volumetric NIfTI image) and how to align it with x (which is already a parcellated DataFrame).
[5]:
from nispace.api import NiSpace
nsp = NiSpace(
x=pet_maps, # reference maps
y=pain_map, # input map — will be parcellated automatically
y_labels="Pain", # a name for our input map
parcellation="Yan200",
n_proc=4, # use 4 parallel processes where possible
seed=42 # for reproducibility
)
print(nsp)
<nispace.api.NiSpace object at 0x103719fd0>
Fitting: parcellation and data ingestion
Calling fit() is where actual data processing begins. For the pain map (a NIfTI image), this means parcellating it into the 200 Yan200 regions. For the PET maps (already a DataFrame), it validates and stores the data.
[6]:
nsp.fit()
# inspect the parcellated pain map
print("Parcellated pain map:")
nsp.get_y()
INFO | 20/07/26 18:19:56 | nispace.api: *** NiSpace.fit() - Data extraction and preparation. ***
INFO | 20/07/26 18:19:56 | nispace.core.parcellation: Building cortex Parcellation for 'Yan200' from library. DOI: 10.1016/j.neuroimage.2023.120010
INFO | 20/07/26 18:19:56 | nispace.core.parcellation: Available spaces: MNI152NLin2009cAsym, MNI152NLin6Asym, fsLR, fsaverage
INFO | 20/07/26 18:19:57 | nispace.core.parcellation: Parcellation 'Yan200': validation passed.
INFO | 20/07/26 18:19:57 | nispace.core.parcellation: Lazy-loading parcellation image for space 'MNI152NLin2009cAsym'.
INFO | 20/07/26 18:19:58 | nispace.core.parcellation: Parcellation 'Yan200': active space set to 'MNI152NLin2009cAsym'.
INFO | 20/07/26 18:19:58 | nispace.api: Checking input data for 'x' (should be, e.g., PET data):
INFO | 20/07/26 18:19:58 | nispace.io: Input type: DataFrame, assuming parcellated data with shape (n_files/subjects/etc, n_parcels).
WARNING | 20/07/26 18:19:58 | nispace.io: Parcellated data contains nan values!
INFO | 20/07/26 18:19:58 | nispace.api: Got 'x' data for 29 x 200 parcels.
INFO | 20/07/26 18:19:58 | nispace.api: Checking input data for 'y' (should be, e.g., subject data):
INFO | 20/07/26 18:19:58 | nispace.io: Input type: list, assuming imaging data.
INFO | 20/07/26 18:19:58 | nispace.io: Background (bg) handling: background_value='auto'; reporting bg-only parcels: False
INFO | 20/07/26 18:19:58 | nispace.io: Parcellating imaging data.
Parcellating (4 proc): 100%|█████████████████████████████████████████████████████████████| 1/1 [00:00<00:00, 116.69it/s]
INFO | 20/07/26 18:20:01 | nispace.api: Got 'y' data for 1 x 200 parcels.
INFO | 20/07/26 18:20:01 | nispace.api: Z-standardizing 'X' data.
Parcellated pain map:
INFO | 20/07/26 18:20:01 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| False |
[6]:
| hemi-L_div-DefaultC_lab-IPL | hemi-L_div-DefaultC_lab-PHC | hemi-L_div-DefaultB_lab-IPL+1 | hemi-L_div-DefaultB_lab-IPL+2 | hemi-L_div-DefaultB_lab-PFCd+1 | hemi-L_div-DefaultB_lab-PFCd+2 | hemi-L_div-DefaultB_lab-PFCl | hemi-L_div-DefaultB_lab-PFCm | hemi-L_div-DefaultB_lab-PFCv+1 | hemi-L_div-DefaultB_lab-PFCv+2 | ... | hemi-R_div-VisualB_lab-ExStrSup | hemi-R_div-VisualB_lab-Striate+1 | hemi-R_div-VisualB_lab-Striate+2 | hemi-R_div-VisualA_lab-ExStr+1 | hemi-R_div-VisualA_lab-ExStr+2 | hemi-R_div-VisualA_lab-ExStr+3 | hemi-R_div-VisualA_lab-ExStr+4 | hemi-R_div-VisualA_lab-SPL | hemi-R_div-VisualA_lab-TempOcc+1 | hemi-R_div-VisualA_lab-TempOcc+2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pain | -1.122654 | -0.286856 | -0.713929 | -1.738277 | -0.458705 | -0.76164 | -1.014422 | -1.082393 | -0.898481 | -0.532955 | ... | -0.811758 | -1.236435 | -1.478081 | -0.750307 | -1.367523 | -0.616786 | -0.443104 | 0.213418 | -0.541916 | -0.464656 |
1 rows × 200 columns
The parcellated data is stored internally. get_y() returns the input maps, get_x() returns the reference maps — both as DataFrames with maps in rows and parcels in columns.
Colocalization
Now for the core step: computing correlations between the pain map and each PET receptor map. We use Spearman rank correlation, which is robust to outliers and doesn’t assume normally distributed data.
[7]:
nsp.colocalize("spearman")
# get the results
colocs = nsp.get_colocalizations()
print(f"Results: {colocs.shape[0]} input maps x {colocs.shape[1]} reference maps")
# show sorted by colocalization value
colocs.T.sort_values(by="Pain", ascending=False)
INFO | 20/07/26 18:20:01 | nispace.api: *** NiSpace.colocalize() - Estimating X & Y colocalizations. ***
INFO | 20/07/26 18:20:01 | nispace.api: Running 'spearman' colocalization.
INFO | 20/07/26 18:20:01 | nispace.api: Pre-ranking X and Y data.
Colocalizing (spearman, 4 proc): 100%|██████████████████████████████████████████████████| 1/1 [00:00<00:00, 2191.38it/s]
INFO | 20/07/26 18:20:04 | nispace.api: Returning colocalizations:
| METHOD | XSEA | X_REDUCTION | Y_TRANSFORM |
| spearman | False | False | False |
Results: 1 input maps x 29 reference maps
[7]:
| Pain | ||
|---|---|---|
| set | map | |
| Noradrenaline/Acetylcholine | target-VAChT_tracer-feobv_n-18_dx-hc_pub-aghourian2017 | 0.422783 |
| Opioids/Endocannabinoids | target-KOR_tracer-ly2795050_n-28_dx-hc_pub-vijay2018 | 0.335397 |
| Noradrenaline/Acetylcholine | target-NET_tracer-mrb_n-10_dx-hc_pub-hesse2017 | 0.288889 |
| Histamine | target-H3_tracer-gsk189254_n-8_dx-hc_pub-gallezot2017 | 0.273213 |
| Glutamate | target-mGluR5_tracer-abp688_n-73_dx-hc_pub-smart2019 | 0.180214 |
| Opioids/Endocannabinoids | target-MOR_tracer-carfentanil_n-204_dx-hc_pub-kantonen2020 | 0.171576 |
| Serotonin | target-5HTT_tracer-dasb_n-18_dx-hc_pub-savli2012 | 0.145715 |
| GABA | target-GABAa5_tracer-ro154513_n-10_dx-hc_pub-lukow2022 | 0.107938 |
| Opioids/Endocannabinoids | target-CB1_tracer-omar_n-77_dx-hc_pub-normandin2015 | 0.101408 |
| Dopamine | target-DAT_tracer-fpcit_n-174_dx-hc_pub-dukart2018 | 0.097589 |
| target-D1_tracer-sch23390_n-13_dx-hc_pub-kaller2017 | 0.083616 | |
| Serotonin | target-5HT1a_tracer-way100635_n-35_dx-hc_pub-savli2012 | 0.063584 |
| Dopamine | target-D23_tracer-flb457_n-55_dx-hc_pub-sandiego2015 | 0.043344 |
| General | target-VMAT2_tracer-dtbz_n-76_dx-hc_pub-larsen2020 | 0.033526 |
| Noradrenaline/Acetylcholine | target-A4B2_tracer-flubatine_n-30_dx-hc_pub-hillmer2016 | 0.030775 |
| Dopamine | target-FDOPA_tracer-fluorodopa_n-12_dx-hc_pub-garciagomez2018 | -0.003416 |
| General | target-SV2A_tracer-ucbj_n-76_dx-hc_pub-finnema2016 | -0.069853 |
| Noradrenaline/Acetylcholine | target-M1_tracer-lsn3172176_n-24_dx-hc_pub-naganawa2020 | -0.104149 |
| Serotonin | target-5HT4_tracer-sb207145_n-59_dx-hc_pub-beliveau2017 | -0.148921 |
| Immunity | target-TSPO_tracer-pbr28_n-6_dx-hc_pub-lois2018 | -0.155657 |
| target-COX1_tracer-ps13_n-11_dx-hc_pub-kim2020 | -0.171675 | |
| Glutamate | target-NMDA_tracer-ge179_n-29_dx-hc_pub-galovic2021 | -0.171878 |
| General | target-HDAC_tracer-martinostat_n-8_dx-hc_pub-wey2016 | -0.179954 |
| target-CMRglu_tracer-fdg_n-20_dx-hc_pub-castrillon2023 | -0.192582 | |
| GABA | target-GABAa_tracer-flumazenil_n-6_dx-hc_pub-dukart2018 | -0.216449 |
| Serotonin | target-5HT6_tracer-gsk215083_n-30_dx-hc_pub-radhakrishnan2018 | -0.272043 |
| target-5HT1b_tracer-p943_n-23_dx-hc_pub-savli2012 | -0.284841 | |
| General | target-rCPS_tracer-leucine_n-42_dx-hc_pub-smith2023 | -0.301752 |
| Serotonin | target-5HT2a_tracer-altanserin_n-19_dx-hc_pub-savli2012 | -0.338328 |
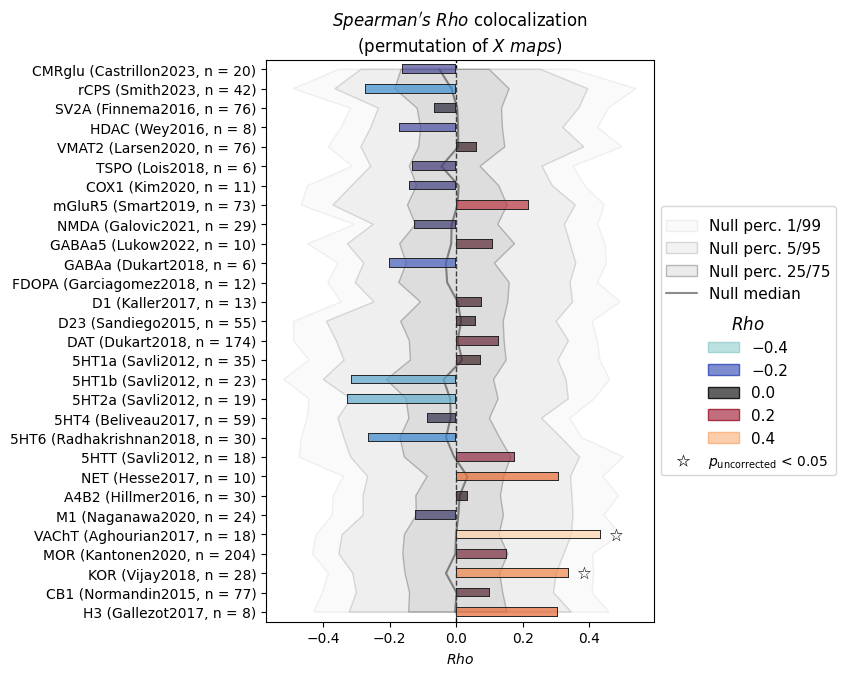
The result is a DataFrame with one row per input map (here: “Pain”) and one column per reference map. The values are Spearman ρ.
We can see which receptors show the strongest positive and negative colocalizations. But these bare correlation values don’t come with reliable p-values yet — we haven’t accounted for spatial autocorrelation. That’s where permutation testing comes in.
Permutation testing
We call permute() with "maps" to generate spatially-constrained null versions of the PET maps and recompute correlations 10,000 times. This gives us an empirical null distribution for each receptor.
We pass p_tails="upper" because our hypothesis is directional: we expect positive colocalizations with receptors involved in pain processing (opioids, noradrenergic/cholinergic pathways).
Null model details are covered in the Null Models notebook. For now, just note that by default NiSpace uses Moran spectral randomization ("moran") for cortical parcellations like Yan200 — it generates surrogate maps that preserve the spatial autocorrelation structure of the original data.
[8]:
nsp.permute(
"maps",
n_perm=1000, # increase to >=10000 for final analyses
p_tails="upper", # one-tailed: test for positive colocalization, default is "two"
)
# multiple comparison correction across the 29 receptor maps
nsp.correct_p() # mc_method argument to specify method, default is "meff" (effective number of tests)
p_values = nsp.get_p_values()
# show p-values sorted ascending
p_values.T.sort_values(by="Pain")
INFO | 20/07/26 18:20:04 | nispace.api: *** NiSpace.permute() - Estimate exact non-parametric p values. ***
INFO | 20/07/26 18:20:04 | nispace.api: Permutation of: X maps.
INFO | 20/07/26 18:20:04 | nispace.api: Using default null method 'moran' (parcellation null space: 'fsLR').
INFO | 20/07/26 18:20:04 | nispace.core.parcellation: Lazy-loading parcellation image for space 'fsLR'.
INFO | 20/07/26 18:20:04 | nispace.api: Loading observed colocalizations (method = 'spearman').
INFO | 20/07/26 18:20:04 | nispace.api: Returning colocalizations:
| METHOD | XSEA | X_REDUCTION | Y_TRANSFORM |
| spearman | False | False | False |
INFO | 20/07/26 18:20:04 | nispace.api: Generating permuted X maps.
INFO | 20/07/26 18:20:04 | nispace.core.permute: Generating null maps (n = 1000, null_method = 'moran').
INFO | 20/07/26 18:20:04 | nispace.nulls: Null map generation: Assuming n = 29 data vector(s) for n = 200 parcels.
INFO | 20/07/26 18:20:04 | nispace.nulls: Using provided distance matrix/matrices.
Moran null maps (4 proc): 100%|█████████████████████████████████████████████████████████| 29/29 [00:00<00:00, 72.19it/s]
INFO | 20/07/26 18:20:07 | nispace.nulls: Null data generation finished.
INFO | 20/07/26 18:20:07 | nispace.core.permute: Z-standardizing null maps.
INFO | 20/07/26 18:20:08 | nispace.api: Pre-ranking X and Y (null) data.
Processing null arrays (4 proc): 100%|█████████████████████████████████████████████| 1000/1000 [00:01<00:00, 827.18it/s]
Null colocalizations (spearman, 4 proc): 100%|████████████████████████████████████| 1000/1000 [00:00<00:00, 7719.57it/s]
INFO | 20/07/26 18:20:09 | nispace.core.permute: Calculating exact p-values (tails = {'rho': 'upper'}).
INFO | 20/07/26 18:20:09 | nispace.api: *** NiSpace.correct_p() - Correct p values for multiple comparisons. ***
INFO | 20/07/26 18:20:09 | nispace.api: Correction method: 'meff_galwey', alpha: 0.05, dimension: 'array'.
INFO | 20/07/26 18:20:09 | nispace.api: Returning X dataframe:
| X_REDUCTION |
| False |
INFO | 20/07/26 18:20:09 | nispace.api: Meff_X (galwey) = 12.16 (from 29 maps).
INFO | 20/07/26 18:20:09 | nispace.api: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | X_REDUCTION | Y_TRANSFORM |
| spearman | xmaps | False | None | False | False |
[8]:
| Pain | ||
|---|---|---|
| set | map | |
| Noradrenaline/Acetylcholine | target-VAChT_tracer-feobv_n-18_dx-hc_pub-aghourian2017 | 0.073 |
| Opioids/Endocannabinoids | target-KOR_tracer-ly2795050_n-28_dx-hc_pub-vijay2018 | 0.103 |
| Histamine | target-H3_tracer-gsk189254_n-8_dx-hc_pub-gallezot2017 | 0.164 |
| Glutamate | target-mGluR5_tracer-abp688_n-73_dx-hc_pub-smart2019 | 0.172 |
| Noradrenaline/Acetylcholine | target-NET_tracer-mrb_n-10_dx-hc_pub-hesse2017 | 0.255 |
| Opioids/Endocannabinoids | target-MOR_tracer-carfentanil_n-204_dx-hc_pub-kantonen2020 | 0.259 |
| Serotonin | target-5HTT_tracer-dasb_n-18_dx-hc_pub-savli2012 | 0.294 |
| Dopamine | target-DAT_tracer-fpcit_n-174_dx-hc_pub-dukart2018 | 0.332 |
| GABA | target-GABAa5_tracer-ro154513_n-10_dx-hc_pub-lukow2022 | 0.347 |
| Opioids/Endocannabinoids | target-CB1_tracer-omar_n-77_dx-hc_pub-normandin2015 | 0.358 |
| Serotonin | target-5HT1a_tracer-way100635_n-35_dx-hc_pub-savli2012 | 0.407 |
| Dopamine | target-D1_tracer-sch23390_n-13_dx-hc_pub-kaller2017 | 0.417 |
| target-D23_tracer-flb457_n-55_dx-hc_pub-sandiego2015 | 0.437 | |
| General | target-VMAT2_tracer-dtbz_n-76_dx-hc_pub-larsen2020 | 0.484 |
| Dopamine | target-FDOPA_tracer-fluorodopa_n-12_dx-hc_pub-garciagomez2018 | 0.502 |
| Noradrenaline/Acetylcholine | target-A4B2_tracer-flubatine_n-30_dx-hc_pub-hillmer2016 | 0.507 |
| Serotonin | target-5HT1b_tracer-p943_n-23_dx-hc_pub-savli2012 | 0.651 |
| Immunity | target-COX1_tracer-ps13_n-11_dx-hc_pub-kim2020 | 0.691 |
| General | target-SV2A_tracer-ucbj_n-76_dx-hc_pub-finnema2016 | 0.779 |
| target-rCPS_tracer-leucine_n-42_dx-hc_pub-smith2023 | 0.786 | |
| Immunity | target-TSPO_tracer-pbr28_n-6_dx-hc_pub-lois2018 | 0.787 |
| General | target-HDAC_tracer-martinostat_n-8_dx-hc_pub-wey2016 | 0.791 |
| target-CMRglu_tracer-fdg_n-20_dx-hc_pub-castrillon2023 | 0.795 | |
| Noradrenaline/Acetylcholine | target-M1_tracer-lsn3172176_n-24_dx-hc_pub-naganawa2020 | 0.795 |
| Serotonin | target-5HT4_tracer-sb207145_n-59_dx-hc_pub-beliveau2017 | 0.804 |
| GABA | target-GABAa_tracer-flumazenil_n-6_dx-hc_pub-dukart2018 | 0.887 |
| Glutamate | target-NMDA_tracer-ge179_n-29_dx-hc_pub-galovic2021 | 0.901 |
| Serotonin | target-5HT6_tracer-gsk215083_n-30_dx-hc_pub-radhakrishnan2018 | 0.931 |
| target-5HT2a_tracer-altanserin_n-19_dx-hc_pub-savli2012 | 0.972 |
After correcting for spatial autocorrelation (via permutation) and for multiple comparisons across the 29 receptor maps (FDR), the cholinergic transporter VAChT stands out with the lowest p-value — consistent with the pain neuroscience literature.
Notice that the effect sizes haven’t changed: permutation testing and correct_p() only affect the p-values, not the correlations.
Visualizing the results
NiSpace has an integrated plot() method that picks a reasonable visualization automatically.
[9]:
nsp.plot()
INFO | 20/07/26 18:20:09 | nispace.api: *** NiSpace.plot() - Plot colocalization results. ***
INFO | 20/07/26 18:20:09 | nispace.api: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | X_REDUCTION | Y_TRANSFORM |
| spearman | xmaps | False | None | False | False |
INFO | 20/07/26 18:20:09 | nispace.api: Returning colocalizations:
| METHOD | XSEA | X_REDUCTION | Y_TRANSFORM |
| spearman | False | False | False |
INFO | 20/07/26 18:20:09 | nispace.api: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | X_REDUCTION | Y_TRANSFORM |
| spearman | xmaps | False | None | False | False |
INFO | 20/07/26 18:20:09 | nispace.api: Returning p values:
| METHOD | PERMUTE_WHAT | XSEA | MC_METHOD | X_REDUCTION | Y_TRANSFORM |
| spearman | xmaps | False | meffgalwey | False | False |
INFO | 20/07/26 18:20:09 | nispace.api: Creating categorical plot for method spearman, colocalization stat rho.
INFO | 20/07/26 18:20:10 | nispace.plotting: Significance annotation: 0/29 p_uncorrected < 0.05, 0/29 p_meffgalwey < 0.05
[9]:
(<Figure size 500x730 with 1 Axes>,
<Axes: title={'center': "$Spearman's\\ Rho$ colocalization\n(permutation of $X\\ maps$)"}, xlabel='$Rho$'>,
<seaborn._core.plot.Plotter at 0x33ea13940>)
We can also visualize the input and reference maps as above, but here directly from NiSpace object:
[10]:
# the pain map
nsp.plot_brain(data="Y")
INFO | 20/07/26 18:20:10 | nispace.api: *** NiSpace.plot_brain() ***
INFO | 20/07/26 18:20:10 | nispace.api: Plotting Y data (Y_transform='False').
INFO | 20/07/26 18:20:10 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| False |
WARNING | 20/07/26 18:20:10 | nispace.plotting: Brain plotting in NiSpace is experimental. If things look off, feel free to raise a GitHub issue!
INFO | 20/07/26 18:20:10 | nispace.plotting: brainplot: threshold='auto' → 0.0017398296622559428
INFO | 20/07/26 18:20:10 | nispace.plotting: brainplot: kind='glass', img_mode='None', surf_space='None', mni_space='MNI152NLin2009cAsym', surf_mesh='inflated'
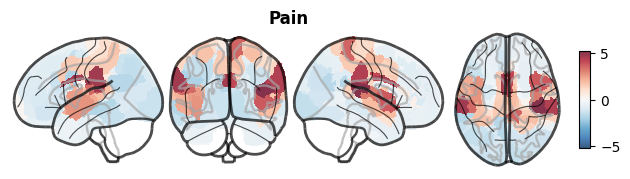
[10]:
(<Figure size 720x180 with 6 Axes>, [<Axes: >])
[11]:
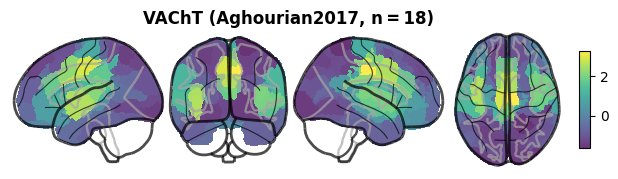
# the VAChT receptor map (strongest colocalization)
nsp.plot_brain(data="X", maps="VAChT", symmetric_cmap=False)
INFO | 20/07/26 18:20:13 | nispace.api: *** NiSpace.plot_brain() ***
INFO | 20/07/26 18:20:13 | nispace.api: Plotting X data (X_reduction='False').
INFO | 20/07/26 18:20:13 | nispace.api: Returning X dataframe:
| X_REDUCTION |
| False |
WARNING | 20/07/26 18:20:13 | nispace.plotting: Brain plotting in NiSpace is experimental. If things look off, feel free to raise a GitHub issue!
INFO | 20/07/26 18:20:13 | nispace.plotting: brainplot: threshold='auto' → 0.0001124086047639139
INFO | 20/07/26 18:20:13 | nispace.plotting: brainplot: kind='glass', img_mode='None', surf_space='None', mni_space='MNI152NLin2009cAsym', surf_mesh='inflated'
[11]:
(<Figure size 720x180 with 6 Axes>, [<Axes: >])
Saving and loading
Analyses with 10,000 permutations or more take time to generate. We can serialize the entire NiSpace object — including the null distributions — to disk with to_pickle(), and load it back with from_pickle().
We recommend the .pkl.blosc format: Blosc compression keeps file sizes manageable without the speed penalty of gzip. When running with many permutations but using a method that generates null maps fast (e.g., "spin" or "moran"), it sometimes makes sense to save without the null maps to spare storage space.
[12]:
import os
# save, including the null distributions
nsp.to_pickle("intro02_nsp.pkl.blosc")
print(f"With nulls: {os.path.getsize('intro02_nsp.pkl.blosc') / 1e6:.1f} MB")
# save without nulls — much smaller, but you lose the shading in plots
nsp.to_pickle("intro02_nsp_no_nulls.pkl.blosc", save_nulls=False)
print(f"Without nulls: {os.path.getsize('intro02_nsp_no_nulls.pkl.blosc') / 1e6:.1f} MB")
With nulls: 166.8 MB
Without nulls: 144.8 MB
[13]:
# load it back
nsp_loaded = NiSpace.from_pickle("intro02_nsp.pkl.blosc")
import numpy as np
assert np.allclose(
nsp.get_colocalizations().values,
nsp_loaded.get_colocalizations().values
)
print("Loaded successfully — results are identical.")
INFO | 20/07/26 18:20:20 | nispace.api: Returning colocalizations:
| METHOD | XSEA | X_REDUCTION | Y_TRANSFORM |
| spearman | False | False | False |
INFO | 20/07/26 18:20:20 | nispace.api: Returning colocalizations:
| METHOD | XSEA | X_REDUCTION | Y_TRANSFORM |
| spearman | False | False | False |
Loaded successfully — results are identical.
Summary
The complete NiSpace workflow in six lines:
nsp = NiSpace(x=reference_maps, y=input_map, parcellation="Yan200")
nsp.fit() # parcellate + ingest
nsp.colocalize("spearman") # compute correlations
nsp.permute("maps", n_perm=10000, p_tails="upper") # permutation p-values
nsp.correct_p(mc_method=...) # multiple comparisons correction
nsp.plot() # visualize
The next notebook covers what datasets and parcellations are available, and how to bring in your own data.
To explore which brain regions drive the colocalization results computed here, see the Regional Influence notebook.