Working with imaging phenotypes (Y data)
In real neuroimaging studies, you rarely have a single group-average brain map ready to go. More often you have individual subject images that you need to transform into a group-level phenotype before you can run a colocalization.
This notebook covers:
Computing group-level effect sizes with
transform_y()(Hedges’ g, Cohen’s d, z-scores, percentiles)Removing confounding effects with
clean_y()(covariate regression, ComBat harmonization)
We use the "anorexianervosa" example dataset: 50 patients and 50 healthy controls with parcellated grey matter values.
Note: This dataset is simulated and not intended for scientific use. The numbers are realistic but fabricated — do not draw any clinical or scientific conclusions from it.
[2]:
import tqdm.notebook
tqdm.notebook.tqdm = tqdm.tqdm
import numpy as np
import pandas as pd
Load the example dataset
The anorexia nervosa example dataset contains parcellated grey matter maps for 100 subjects. The group labels are encoded in the subject IDs.
[3]:
from nispace.datasets import fetch_example, fetch_reference
# load individual subject data
an_data = fetch_example("anorexianervosa", parcellation="Yan200")
# extract group labels from subject IDs
groups = an_data.index.str.extract(r'(AN|HC)$')[0].values
print(f"Data: {an_data.shape[0]} subjects x {an_data.shape[1]} parcels")
print(f"Groups: {pd.Series(groups).value_counts().to_dict()}")
an_data.head(3)
INFO | 20/07/26 18:20:36 | nispace.datasets: Loading example dataset: 'anorexianervosa', parcellated with: Yan200.
Data: 100 subjects x 200 parcels
Groups: {'AN': 50, 'HC': 50}
[3]:
| hemi-L_div-DefaultC_lab-IPL | hemi-L_div-DefaultC_lab-PHC | hemi-L_div-DefaultB_lab-IPL+1 | hemi-L_div-DefaultB_lab-IPL+2 | hemi-L_div-DefaultB_lab-PFCd+1 | hemi-L_div-DefaultB_lab-PFCd+2 | hemi-L_div-DefaultB_lab-PFCl | hemi-L_div-DefaultB_lab-PFCm | hemi-L_div-DefaultB_lab-PFCv+1 | hemi-L_div-DefaultB_lab-PFCv+2 | ... | hemi-R_div-VisualB_lab-ExStrSup | hemi-R_div-VisualB_lab-Striate+1 | hemi-R_div-VisualB_lab-Striate+2 | hemi-R_div-VisualA_lab-ExStr+1 | hemi-R_div-VisualA_lab-ExStr+2 | hemi-R_div-VisualA_lab-ExStr+3 | hemi-R_div-VisualA_lab-ExStr+4 | hemi-R_div-VisualA_lab-SPL | hemi-R_div-VisualA_lab-TempOcc+1 | hemi-R_div-VisualA_lab-TempOcc+2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sub-001AN | 0.419538 | 0.668976 | 0.558926 | 0.413274 | 0.450007 | 0.516978 | 0.462198 | 0.608400 | 0.506976 | 0.711472 | ... | 0.528548 | 0.595074 | 0.676901 | 0.437711 | 0.521024 | 0.588184 | 0.672914 | 0.395552 | 0.575377 | 0.656451 |
| sub-002AN | 0.592581 | 0.562650 | 0.519997 | 0.570011 | 0.560273 | 0.582357 | 0.306417 | 0.489627 | 0.448591 | 0.412901 | ... | 0.714610 | 0.477251 | 0.422106 | 0.501309 | 0.555876 | 0.603978 | 0.732074 | 0.231209 | 0.638542 | 0.571437 |
| sub-003AN | 0.534957 | 0.716447 | 0.566856 | 0.567441 | 0.433479 | 0.453510 | 0.502837 | 0.569255 | 0.520615 | 0.645931 | ... | 0.653850 | 0.609520 | 0.560292 | 0.601528 | 0.604868 | 0.532327 | 0.782942 | 0.370321 | 0.513307 | 0.581334 |
3 rows × 200 columns
Setting up NiSpace with individual subject data
When y is a DataFrame with one row per subject, NiSpace treats it as individual-level data. We pass the group vector separately when we call transform_y().
We also load the PET reference data.
[4]:
from nispace.api import NiSpace
pet_maps = fetch_reference("pet", parcellation="Yan200",
collection="UniqueTracers", print_references=False)
nsp = NiSpace(
x=pet_maps,
y=an_data, # individual subject maps — one row per subject
parcellation="Yan200",
n_proc=4,
seed=42
)
nsp.fit()
print(f"Fitted. Y shape: {nsp.get_y().shape}")
INFO | 20/07/26 18:20:36 | nispace.datasets: Loading pet maps.
INFO | 20/07/26 18:20:36 | nispace.datasets: Loading integrated collection 'UniqueTracers' for dataset 'pet'.
INFO | 20/07/26 18:20:36 | nispace.datasets: Filtering maps by collection.
INFO | 20/07/26 18:20:36 | nispace.datasets: Loading data parcellated with 'Yan200'
INFO | 20/07/26 18:20:36 | nispace.api: *** NiSpace.fit() - Data extraction and preparation. ***
INFO | 20/07/26 18:20:36 | nispace.core.parcellation: Building cortex Parcellation for 'Yan200' from library. DOI: 10.1016/j.neuroimage.2023.120010
INFO | 20/07/26 18:20:36 | nispace.core.parcellation: Available spaces: MNI152NLin2009cAsym, MNI152NLin6Asym, fsLR, fsaverage
INFO | 20/07/26 18:20:37 | nispace.core.parcellation: Parcellation 'Yan200': validation passed.
INFO | 20/07/26 18:20:37 | nispace.api: Checking input data for 'x' (should be, e.g., PET data):
INFO | 20/07/26 18:20:37 | nispace.io: Input type: DataFrame, assuming parcellated data with shape (n_files/subjects/etc, n_parcels).
WARNING | 20/07/26 18:20:37 | nispace.io: Parcellated data contains nan values!
INFO | 20/07/26 18:20:37 | nispace.api: Got 'x' data for 29 x 200 parcels.
INFO | 20/07/26 18:20:37 | nispace.api: Checking input data for 'y' (should be, e.g., subject data):
INFO | 20/07/26 18:20:37 | nispace.io: Input type: DataFrame, assuming parcellated data with shape (n_files/subjects/etc, n_parcels).
INFO | 20/07/26 18:20:37 | nispace.api: Got 'y' data for 100 x 200 parcels.
INFO | 20/07/26 18:20:37 | nispace.api: Z-standardizing 'X' data.
INFO | 20/07/26 18:20:37 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| False |
Fitted. Y shape: (100, 200)
Computing group-level effect sizes with transform_y()
To run a spatial colocalization, we need a single group-level map. transform_y() computes this for us, using a formula that specifies the comparison.
The group vector tells NiSpace which subjects belong to which group. The convention is that group “a” (here: AN) is compared against group “b” (here: HC).
Hedges’ g
Hedges’ g is a bias-corrected version of Cohen’s d — the most common effect size metric for group comparisons. A negative g at a given parcel means the AN group has lower grey matter there compared to controls.
[5]:
# compute Hedges' g: AN vs. HC
# the formula "hedges(a,b)" computes Hedges' g for group a (AN) vs. group b (HC)
nsp.transform_y(
transform="hedges(a,b)",
groups=groups # vector of group labels, same length as number of subjects
)
hedges_g = nsp.get_y()
print(f"After transform_y: {hedges_g.shape} (one row = one group comparison)")
print(f"Value range: [{hedges_g.values.min():.2f}, {hedges_g.values.max():.2f}]")
hedges_g
INFO | 20/07/26 18:20:37 | nispace.api: *** NiSpace.transform_y() - Y transformation and comparison. ***
INFO | 20/07/26 18:20:37 | nispace.api: Groups/sessions vector provided, ensuring dummy-coding.
INFO | 20/07/26 18:20:37 | nispace.api: Applying Y transform 'hedges(a,b)'.
INFO | 20/07/26 18:20:37 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| hedges(a,b) |
After transform_y: (1, 200) (one row = one group comparison)
Value range: [-0.27, 0.86]
[5]:
| hemi-L_div-DefaultC_lab-IPL | hemi-L_div-DefaultC_lab-PHC | hemi-L_div-DefaultB_lab-IPL+1 | hemi-L_div-DefaultB_lab-IPL+2 | hemi-L_div-DefaultB_lab-PFCd+1 | hemi-L_div-DefaultB_lab-PFCd+2 | hemi-L_div-DefaultB_lab-PFCl | hemi-L_div-DefaultB_lab-PFCm | hemi-L_div-DefaultB_lab-PFCv+1 | hemi-L_div-DefaultB_lab-PFCv+2 | ... | hemi-R_div-VisualB_lab-ExStrSup | hemi-R_div-VisualB_lab-Striate+1 | hemi-R_div-VisualB_lab-Striate+2 | hemi-R_div-VisualA_lab-ExStr+1 | hemi-R_div-VisualA_lab-ExStr+2 | hemi-R_div-VisualA_lab-ExStr+3 | hemi-R_div-VisualA_lab-ExStr+4 | hemi-R_div-VisualA_lab-SPL | hemi-R_div-VisualA_lab-TempOcc+1 | hemi-R_div-VisualA_lab-TempOcc+2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| hedges | 0.465267 | -0.1308 | 0.098488 | 0.145487 | 0.848914 | 0.682891 | 0.143101 | 0.456289 | 0.227216 | -0.024675 | ... | 0.400688 | 0.041456 | 0.252205 | 0.08656 | 0.060542 | -0.116548 | -0.114368 | 0.516329 | 0.523495 | 0.322697 |
1 rows × 200 columns
[6]:
# visualize on the brain
nsp.plot_brain(data="Y", symmetric_cmap=True)
INFO | 20/07/26 18:20:37 | nispace.api: *** NiSpace.plot_brain() ***
INFO | 20/07/26 18:20:37 | nispace.api: Plotting Y data (Y_transform='hedges(a,b)').
INFO | 20/07/26 18:20:37 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| hedges(a,b) |
WARNING | 20/07/26 18:20:37 | nispace.plotting: Brain plotting in NiSpace is experimental. If things look off, feel free to raise a GitHub issue!
INFO | 20/07/26 18:20:37 | nispace.plotting: brainplot: threshold='auto' → 0.00015760881069581956
INFO | 20/07/26 18:20:37 | nispace.core.parcellation: Lazy-loading parcellation image for space 'MNI152NLin2009cAsym'.
INFO | 20/07/26 18:20:37 | nispace.core.parcellation: Parcellation 'Yan200': active space set to 'MNI152NLin2009cAsym'.
INFO | 20/07/26 18:20:37 | nispace.plotting: brainplot: kind='glass', img_mode='None', surf_space='None', mni_space='MNI152NLin2009cAsym', surf_mesh='inflated'
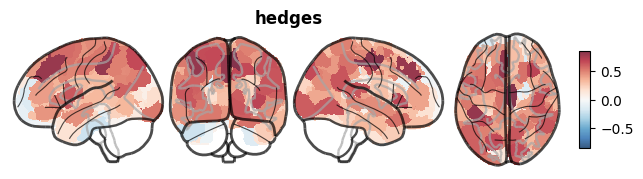
[6]:
(<Figure size 720x180 with 6 Axes>, [<Axes: >])
Widespread negative values are expected: anorexia nervosa is associated with global grey matter reductions, most pronounced in frontal and temporal regions.
Cohen’s d
Cohen’s d is the non-bias-corrected version. With n=50 per group the difference from Hedges’ g is minimal, but it’s good to know both are available.
[7]:
nsp.transform_y(transform="cohen(a,b)", groups=groups)
cohens_d = nsp.get_y()
print(f"Cohen's d range: [{cohens_d.values.min():.2f}, {cohens_d.values.max():.2f}]")
INFO | 20/07/26 18:20:41 | nispace.api: *** NiSpace.transform_y() - Y transformation and comparison. ***
INFO | 20/07/26 18:20:41 | nispace.api: Groups/sessions vector provided, ensuring dummy-coding.
INFO | 20/07/26 18:20:41 | nispace.api: Applying Y transform 'cohen(a,b)'.
INFO | 20/07/26 18:20:41 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| cohen(a,b) |
Cohen's d range: [-0.27, 0.87]
Z-scores (individual deviation maps)
Instead of a single group map, we can compute a deviation map for each patient: how many standard deviations does this individual deviate from the control mean at each parcel? This is useful for individual-level analyses — see Notebook 11.
[8]:
# z-score: each AN subject mapped relative to HC mean and SD
nsp.transform_y(transform="zscore(a,b)", groups=groups)
zscores = nsp.get_y()
print(f"Z-score maps: {zscores.shape} (one row per AN subject)")
zscores.head(3)
INFO | 20/07/26 18:20:41 | nispace.api: *** NiSpace.transform_y() - Y transformation and comparison. ***
INFO | 20/07/26 18:20:41 | nispace.api: Groups/sessions vector provided, ensuring dummy-coding.
INFO | 20/07/26 18:20:41 | nispace.api: Applying Y transform 'zscore(a,b)'.
INFO | 20/07/26 18:20:41 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| zscore(a,b) |
Z-score maps: (50, 200) (one row per AN subject)
[8]:
| hemi-L_div-DefaultC_lab-IPL | hemi-L_div-DefaultC_lab-PHC | hemi-L_div-DefaultB_lab-IPL+1 | hemi-L_div-DefaultB_lab-IPL+2 | hemi-L_div-DefaultB_lab-PFCd+1 | hemi-L_div-DefaultB_lab-PFCd+2 | hemi-L_div-DefaultB_lab-PFCl | hemi-L_div-DefaultB_lab-PFCm | hemi-L_div-DefaultB_lab-PFCv+1 | hemi-L_div-DefaultB_lab-PFCv+2 | ... | hemi-R_div-VisualB_lab-ExStrSup | hemi-R_div-VisualB_lab-Striate+1 | hemi-R_div-VisualB_lab-Striate+2 | hemi-R_div-VisualA_lab-ExStr+1 | hemi-R_div-VisualA_lab-ExStr+2 | hemi-R_div-VisualA_lab-ExStr+3 | hemi-R_div-VisualA_lab-ExStr+4 | hemi-R_div-VisualA_lab-SPL | hemi-R_div-VisualA_lab-TempOcc+1 | hemi-R_div-VisualA_lab-TempOcc+2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sub-001AN | -0.649976 | 0.772236 | 1.121429 | -1.492332 | -0.380535 | 1.004667 | -0.072329 | 0.778237 | -0.535533 | 1.105893 | ... | 0.132797 | -0.203132 | 1.850587 | -0.789601 | -0.798701 | 0.905963 | -0.337266 | 0.676954 | 0.266444 | 0.435931 |
| sub-002AN | 1.273077 | -0.257376 | 0.699317 | 0.225262 | 0.747683 | 1.701899 | -1.503932 | -0.438771 | -1.234989 | -2.334603 | ... | 2.274353 | -1.522535 | -0.708592 | -0.114546 | -0.424343 | 1.089020 | 0.337359 | -1.190730 | 0.975135 | -0.474356 |
| sub-003AN | 0.632687 | 1.231919 | 1.207417 | 0.197090 | -0.549648 | 0.327815 | 0.301143 | 0.377142 | -0.372144 | 0.350650 | ... | 1.575005 | -0.041360 | 0.679359 | 0.949228 | 0.101898 | 0.258560 | 0.917431 | 0.390216 | -0.429965 | -0.368381 |
3 rows × 200 columns
Centile scores (group-relative percentile rank)
Centile scores place each patient on the normative percentile scale at each parcel: a value of 10 at a given parcel means the patient’s grey matter there is lower than 90% of healthy controls. This is a normative modeling approach useful for characterizing individual patients against a reference population.
Like z-scores, this produces one map per patient — useful for individual-level analyses.
[9]:
# centile: each AN subject ranked within HC distribution
nsp.transform_y(transform="centile(a,b)", groups=groups)
centiles = nsp.get_y()
print(f"Centile maps: {centiles.shape} (one row per AN subject)")
print(f"Value range: [{centiles.values.min():.1f}, {centiles.values.max():.1f}] (percentile, 0–100)")
centiles.head(3)
INFO | 20/07/26 18:20:41 | nispace.api: *** NiSpace.transform_y() - Y transformation and comparison. ***
INFO | 20/07/26 18:20:41 | nispace.api: Groups/sessions vector provided, ensuring dummy-coding.
INFO | 20/07/26 18:20:41 | nispace.api: Applying Y transform 'centile(a,b)'.
INFO | 20/07/26 18:20:41 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| centile(a,b) |
Centile maps: (50, 200) (one row per AN subject)
Value range: [0.0, 100.0] (percentile, 0–100)
[9]:
| hemi-L_div-DefaultC_lab-IPL | hemi-L_div-DefaultC_lab-PHC | hemi-L_div-DefaultB_lab-IPL+1 | hemi-L_div-DefaultB_lab-IPL+2 | hemi-L_div-DefaultB_lab-PFCd+1 | hemi-L_div-DefaultB_lab-PFCd+2 | hemi-L_div-DefaultB_lab-PFCl | hemi-L_div-DefaultB_lab-PFCm | hemi-L_div-DefaultB_lab-PFCv+1 | hemi-L_div-DefaultB_lab-PFCv+2 | ... | hemi-R_div-VisualB_lab-ExStrSup | hemi-R_div-VisualB_lab-Striate+1 | hemi-R_div-VisualB_lab-Striate+2 | hemi-R_div-VisualA_lab-ExStr+1 | hemi-R_div-VisualA_lab-ExStr+2 | hemi-R_div-VisualA_lab-ExStr+3 | hemi-R_div-VisualA_lab-ExStr+4 | hemi-R_div-VisualA_lab-SPL | hemi-R_div-VisualA_lab-TempOcc+1 | hemi-R_div-VisualA_lab-TempOcc+2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sub-001AN | 24.0 | 80.0 | 90.0 | 8.0 | 36.0 | 84.0 | 44.0 | 82.0 | 26.0 | 82.0 | ... | 52.0 | 44.0 | 94.0 | 26.0 | 20.0 | 80.0 | 42.0 | 76.0 | 64.0 | 68.0 |
| sub-002AN | 90.0 | 36.0 | 82.0 | 62.0 | 74.0 | 98.0 | 6.0 | 34.0 | 8.0 | 0.0 | ... | 100.0 | 10.0 | 22.0 | 46.0 | 38.0 | 88.0 | 68.0 | 14.0 | 82.0 | 36.0 |
| sub-003AN | 74.0 | 90.0 | 90.0 | 60.0 | 22.0 | 68.0 | 62.0 | 68.0 | 36.0 | 64.0 | ... | 94.0 | 52.0 | 78.0 | 78.0 | 58.0 | 62.0 | 80.0 | 64.0 | 34.0 | 40.0 |
3 rows × 200 columns
For colocalization analyses, we typically average across subjects to get one group-level map. The Hedges’ g approach does this in one step. But for individual-level colocalization you’d keep all rows and analyze them separately.
Removing confounds with clean_y()
Grey matter maps are affected by confounds that have nothing to do with the disorder: age, sex, total intracranial volume, and scanner site are the usual suspects. clean_y() lets you regress these out before running colocalization.
NiSpace distinguishes between two types of regression:
“within”: regression across parcels within each map (e.g., regressing out a whole-brain grey matter map to control for global effects)
“between”: regression across subjects (e.g., removing age or sex effects)
Let’s generate a synthetic age vector for demonstration and show how to regress it out.
[10]:
# reset to individual subject data for this demo
nsp_clean = NiSpace(
x=pet_maps,
y=an_data,
parcellation="Yan200",
seed=42
)
nsp_clean.fit()
# generate a synthetic age vector: AN mean=25, HC mean=30, SD=5 for both
# (realistic for this kind of study)
rng = np.random.default_rng(42)
age_AN = rng.normal(25, 5, 50)
age_HC = rng.normal(30, 5, 50)
age = np.concatenate([age_AN, age_HC])
# sex: roughly balanced (0=male, 1=female), NiSpace treats string covariates automatically as categorical
sex = rng.permutation(["f"]*80 + ["m"]*20)
print(f"Age: mean={age.mean():.1f}, SD={age.std():.1f}")
print(f"Sex: {(sex=='f').sum()} female, {(sex=='m').sum()} male")
INFO | 20/07/26 18:20:41 | nispace.api: *** NiSpace.fit() - Data extraction and preparation. ***
INFO | 20/07/26 18:20:41 | nispace.core.parcellation: Building cortex Parcellation for 'Yan200' from library. DOI: 10.1016/j.neuroimage.2023.120010
INFO | 20/07/26 18:20:41 | nispace.core.parcellation: Available spaces: MNI152NLin2009cAsym, MNI152NLin6Asym, fsLR, fsaverage
INFO | 20/07/26 18:20:42 | nispace.core.parcellation: Parcellation 'Yan200': validation passed.
INFO | 20/07/26 18:20:42 | nispace.api: Checking input data for 'x' (should be, e.g., PET data):
INFO | 20/07/26 18:20:42 | nispace.io: Input type: DataFrame, assuming parcellated data with shape (n_files/subjects/etc, n_parcels).
WARNING | 20/07/26 18:20:42 | nispace.io: Parcellated data contains nan values!
INFO | 20/07/26 18:20:42 | nispace.api: Got 'x' data for 29 x 200 parcels.
INFO | 20/07/26 18:20:42 | nispace.api: Checking input data for 'y' (should be, e.g., subject data):
INFO | 20/07/26 18:20:42 | nispace.io: Input type: DataFrame, assuming parcellated data with shape (n_files/subjects/etc, n_parcels).
INFO | 20/07/26 18:20:42 | nispace.api: Got 'y' data for 100 x 200 parcels.
INFO | 20/07/26 18:20:42 | nispace.api: Z-standardizing 'X' data.
Age: mean=27.2, SD=4.2
Sex: 80 female, 20 male
[11]:
# regress age and sex out from the individual subject maps
# how="between" means: regression across subjects
# covariates_between is a DataFrame with one row per subject
covariates = pd.DataFrame({"age": age, "sex": sex}, index=an_data.index)
nsp_clean.clean_y(
how="between",
covariates_between=covariates
)
print("Covariate regression done.")
print(f"Y shape after cleaning: {nsp_clean.get_y().shape}")
INFO | 20/07/26 18:20:42 | nispace.api: *** NiSpace.clean_y() - Y covariate regression. ***
INFO | 20/07/26 18:20:42 | nispace.api: Performing covariate regression between maps/subjects (e.g., age, sex, site).
INFO | 20/07/26 18:20:42 | nispace.core.clean_y: Detected categorical covariates: ['sex']; continuous: ['age'].
INFO | 20/07/26 18:20:42 | nispace.core.clean_y: Regressing 2 between covariate(s) from Y.
Regressing 2 between covariate(s) from Y (1 proc): 100%|███████████████████████████| 200/200 [00:00<00:00, 15658.28it/s]
Covariate regression done.
INFO | 20/07/26 18:20:42 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| False |
Y shape after cleaning: (100, 200)
After cleaning, we can compute effect sizes on the covariate-corrected maps:
[12]:
nsp_clean.transform_y(transform="hedges(a,b)", groups=groups)
hedges_g_clean = nsp_clean.get_y()
print(f"Hedges' g after covariate regression: range [{hedges_g_clean.values.min():.2f}, {hedges_g_clean.values.max():.2f}]")
INFO | 20/07/26 18:20:42 | nispace.api: *** NiSpace.transform_y() - Y transformation and comparison. ***
INFO | 20/07/26 18:20:42 | nispace.api: Groups/sessions vector provided, ensuring dummy-coding.
INFO | 20/07/26 18:20:42 | nispace.api: Applying Y transform 'hedges(a,b)'.
INFO | 20/07/26 18:20:42 | nispace.api: Returning Y dataframe:
| Y_TRANSFORM |
| hedges(a,b) |
Hedges' g after covariate regression: range [-0.21, 0.87]
ComBat harmonization
If your data comes from multiple scanners or study sites, clean_y() also supports ComBat harmonization (a Bayesian framework for removing scanner effects while preserving biological variability). Pass combat=True and include a "site" column in your covariates DataFrame:
covariates_with_site = pd.DataFrame({
"age": age,
"sex": sex,
"site": site_labels # scanner/site identifier per subject
}, index=an_data.index)
nsp_clean.clean_y(
how="between",
covariates_between=covariates_with_site,
combat=True
)
ComBat removes site effects while keeping the biological covariates (age, sex) protected.
The full preprocessing pipeline
Putting it all together, a typical preprocessing pipeline before colocalization looks like this:
nsp = NiSpace(x=pet_maps, y=an_data, parcellation="Yan200")
nsp.fit() # parcellate
nsp.clean_y( # remove confounds
how="between",
covariates_between=covariates
)
nsp.transform_y( # compute effect sizes
transform="hedges(a,b)",
groups=groups
)
nsp.colocalize("spearman") # correlate with reference
nsp.permute("groups", n_perm=10000) # permutation test (group labels)
nsp.plot()
Note that when group labels are permuted (permute("groups")), we permute the subject labels before computing the effect size — this gives a more principled null model than permuting the maps. This is covered in detail in Notebook 5.
Summary
Method |
What it does |
|---|---|
|
Hedges’ g per parcel (bias-corrected effect size, group a vs. b) |
|
Cohen’s d per parcel |
|
Z-score per subject (group a relative to b distribution) |
|
Centile score per subject (percentile rank within group b) |
|
Regress out between-subject confounds |
|
Regress out within-map confounds (e.g., grey matter) |
|
ComBat site harmonization |
Next: Notebook 5 covers the null model methods in detail.